Chapter 1: An introduction to basic Ruffus syntax¶
See also
Overview¶
Computational pipelines transform your data in stages until the final result is produced. One easy way to understand pipelines is by imagining your data flowing across a series of pipes until it reaches its final destination. Even quite complicated processes can be broken into simple stages. Of course, it helps to visualise the whole process.
Ruffus is a way of automating the plumbing in your pipeline: You supply the python functions which perform the data transformation, and tell Ruffus how these pipeline task functions are connected up. Ruffus will make sure that the right data flows down your pipeline in the right way at the right time.
Note
Ruffus refers to each stage of your pipeline as a task.
Importing Ruffus¶
The most convenient way to use Ruffus is to import the various names directly:
from ruffus import *This will allow Ruffus terms to be used directly in your code. This is also the style we have adopted for this manual.
- If any of these clash with names in your code, you can use qualified names instead:
Ruffus uses only standard python syntax.
There is no need to install anything extra or to have your script “preprocessed” to run your pipeline.
Ruffus decorators¶
To let Ruffus know that which python functions are part of your pipeline, they need to be tagged or annotated using Ruffus decorators .
Decorators have been part of the Python language since version 2.4. Common examples from the standard library include @staticmethod and classmethod.
decorators start with a @ prefix, and take a number of parameters in parenthesis, much like in a function call.
decorators are placed before a normal python function.
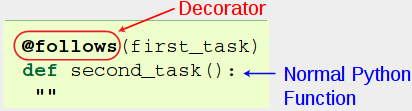
Multiple decorators can be stacked as necessary in whichever order:
@follows(first_task) @follows(another_task) @originate(range(5)) def second_task(): ""Ruffus decorators do not otherwise alter the underlying function. These can still be called normally.
Your first Ruffus pipeline¶
1. Write down the file names¶
Ruffus is designed for data moving through a computational pipeline as a series of files.
It is also possible to use Ruffus pipelines without using intermediate data files but for your first efforts, it is probably best not to subvert its canonical design.
The first thing when designing a new Ruffus pipeline is to sketch out the set of file names for the pipeline on paper:

- Here we have a number of DNA sequence files (*.fasta)
- mapped to a genome (*.sam), and
- compressed (*.bam) before being
- summarised statistically (*.statistics)
The first striking thing is that all of the files following the same consistent naming scheme.
Note
The most important part of a Ruffus pipeline is to have a consistent naming scheme for your files.
This allows you to build sane pipelines.
In this case, each of the files at the same stage share the same file extension, e.g. (.sam). This is usually the simplest and most sensible choice. (We shall see in later chapters that Ruffus supports more complicated naming patterns so long as they are consistent.)
2. Write the python functions for each stage¶
Next, we can sketch out the python functions which do the actual work for the pipeline.
Note
These are normal python functions with the important proviso that
- The first parameter contains the Input (file names)
- The second parameter contains the Output (file names)
You can otherwise supply as many parameters as is required.
Each python function should only take a Single Input at a time
All the parallelism in your pipeline should be handled by Ruffus. Make sure each function analyses one thing at a time.
Ruffus refers to a pipelined function as a task.
The code for our three task functions look something like:
# # STAGE 1 fasta->sam # def map_dna_sequence(input_file, # 1st parameter is Input output_file): # 2nd parameter is Output """ Sketch of real mapping function We can do the mapping ourselves or call some other programme: os.system("stampy %s %s..." % (input_file, output_file)) """ ii = open(input_file) oo = open(output_file, "w")# # STAGE 2 sam->bam # def compress_sam_file(input_file, # Input parameter output_file): # Output parameter """ Sketch of real compression function """ ii = open(input_file) oo = open(output_file, "w")# # STAGE 3 bam->statistics # def summarise_bam_file(input_file, # Input parameter output_file, # Output parameter extra_stats_parameter): # Any number of extra parameters as required """ Sketch of real analysis function """ ii = open(input_file) oo = open(output_file, "w")If we were calling our functions manually, without the benefit of Ruffus, we would need the following sequence of calls:
# STAGE 1 map_dna_sequence("a.fasta", "a.sam") map_dna_sequence("b.fasta", "b.sam") map_dna_sequence("c.fasta", "c.sam") # STAGE 2 compress_sam_file("a.sam", "a.bam") compress_sam_file("b.sam", "b.bam") compress_sam_file("c.sam", "c.bam") # STAGE 3 summarise_bam_file("a.bam", "a.statistics") summarise_bam_file("b.bam", "b.statistics") summarise_bam_file("c.bam", "c.statistics")
3. Link the python functions into a pipeline¶
Ruffus makes exactly the same function calls on your behalf. However, first, we need to tell Ruffus what the arguments should be for each of the function calls.
- The Input is easy: This is either the starting file set (*.fasta) or whatever is produced by the previous stage.
- The Output file name is the same as the Input but with the appropriate extension.
These are specified using the Ruffus @transform decorator as follows:
from ruffus import * starting_files = ["a.fasta", "b.fasta", "c.fasta"] # # STAGE 1 fasta->sam # @transform(starting_files, # Input = starting files suffix(".fasta"), # suffix = .fasta ".sam") # Output suffix = .sam def map_dna_sequence(input_file, output_file): ii = open(input_file) oo = open(output_file, "w") # # STAGE 2 sam->bam # @transform(map_dna_sequence, # Input = previous stage suffix(".sam"), # suffix = .sam ".bam") # Output suffix = .bam def compress_sam_file(input_file, output_file): ii = open(input_file) oo = open(output_file, "w") # # STAGE 3 bam->statistics # @transform(compress_sam_file, # Input = previous stage suffix(".bam"), # suffix = .bam ".statistics", # Output suffix = .statistics "use_linear_model") # Extra statistics parameter def summarise_bam_file(input_file, output_file, extra_stats_parameter): """ Sketch of real analysis function """ ii = open(input_file) oo = open(output_file, "w")
4. @transform syntax¶
Other parameters can be passed to @transform and they will be forwarded to our python pipeline function.
The functions that do the actual work of each stage of the pipeline remain unchanged. The role of Ruffus is to make sure each is called in the right order, with the right parameters, running in parallel (using multiprocessing if desired).
5. Run the pipeline!¶
Note
Key Ruffus Terminology:
A task is an annotated python function which represents a recipe or stage of your pipeline.
A job is each time your recipe is applied to a piece of data, i.e. each time Ruffus calls your function.
Each task or pipeline recipe can thus have many jobs each of which can work in parallel on different data.
Now we can run the pipeline with the Ruffus function pipeline_run:
pipeline_run()This produces three sets of results in parallel, as you might expect:
>>> pipeline_run() Job = [a.fasta -> a.sam] completed Job = [b.fasta -> b.sam] completed Job = [c.fasta -> c.sam] completed Completed Task = map_dna_sequence Job = [a.sam -> a.bam] completed Job = [b.sam -> b.bam] completed Job = [c.sam -> c.bam] completed Completed Task = compress_sam_file Job = [a.bam -> a.statistics, use_linear_model] completed Job = [b.bam -> b.statistics, use_linear_model] completed Job = [c.bam -> c.statistics, use_linear_model] completed Completed Task = summarise_bam_fileTo work out which functions to call, pipeline_run finds the last task function of your pipeline, then works out all the other functions this depends on, working backwards up the chain of dependencies automatically.
We can specify this end point of your pipeline explicitly:
>>> pipeline_run(target_tasks = [summarise_bam_file])This allows us to only run part of the pipeline, for example:
>>> pipeline_run(target_tasks = [compress_sam_file])
Note
The example code can be copied and pasted into a python command shell.